How to use NMR to solve 3D molecule structures
15 Apr 2023There are a few steps to get 3D molecule structures from NMR data:
- Assign NMR peaks to atoms.
- Use NOE and ROE data to get the distance between protons.
- Use Molecular dynamics to calculate the 3D structure based on the distance information.
2 Dimensional NMR Experiments
There are a few popular 2 Dimensional NMR Experiments. Depending on whether both dimensions are on the same type of elements, we can call them homonuclear 2D NMR experiments and heteronuclear 2D NMR experiments.
Homonuclear 2-Dimensional NMR Experiments
Homonuclear 2-Dimensional NMR Experiments are mostly done for protons.
Correlation Spectroscopy (COSY)
COSY NMR spectrum will show peaks for neighboring protons, like the two protons in H-N-C-H or H-C-C-H.
The blue dots in the images show the COSY peaks:
Total correlation spectroscopy(TOCSY)
The TOCSY experiments will show peaks for protons in the same spin system. In the above link, the blue and red peaks together show all the TOCSY peaks for amino acids.
NOTE: for molecules in specific size ranges, the TOCSY experiments will show some ROESY peaks.
Nuclear Overhauser Effect Spectroscopy(NOESY)
The protons locate less than 5Å will show NOESY peaks.
Based on the signal strength(indicated by integration of the peaks or the number of contours), we can compare the distance of protons and categorize them into 1.7-2.5, 2.5-3.5, 3.5-4.5, and 4.5-5.5 ranges.
Heteronuclear 2D NMR experiments
Heteronuclear 2d NMR experiments are usually performed between proton and Carbon 13 or Nitrogen 15.
Heteronuclear single-quantum correlation spectroscopy (HSQC)
HSQC show peaks for directly connected atoms. C13 HSQC will show peaks for H-C bond and N15 HSQC will show peaks for H-N bond.
Heteronuclear multiple-bond correlation spectroscopy (HMBC)
HMBC show peaks for atoms between 2-4 bonds. C13 HMBC will show peaks for H-C-C, H-C-C-C or H-C-C-C-C.
Molecular dynamics
After we get the NOE peak, we can convert them to distance ranges. Then we could use molecular dynamics with those ranges to calculate 3D molecule structures. The software I have used is MacroModel in Schrödinger Suite.
The steps to perform the Molecular Dynamics calculation are:
- Copy the structure from ChemDraw to Maestro.
- The structure is copied from 2D canvas to 3D Canvas. So please do a small relaxation (minimization or molecular dynamics) for the molecule.
- Sometimes the chirality of the molecule may be wrong after the conversion.
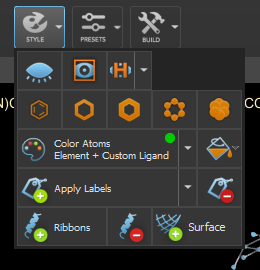
- Check all the chiral atoms and correct them if they are wrong. There is style -> apply labels -> charity to label them automatically.
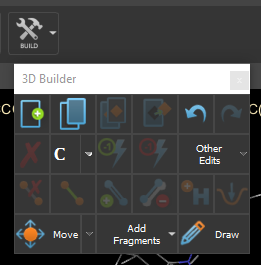
- Sometimes, you could use build -> other edits -> invert chirality to fix them. Other times you have to use the 3d builder panel to remove and redraw hydrogen. I hope those 2 methods help you to fix it.
- Check all the chiral atoms and correct them if they are wrong. There is style -> apply labels -> charity to label them automatically.
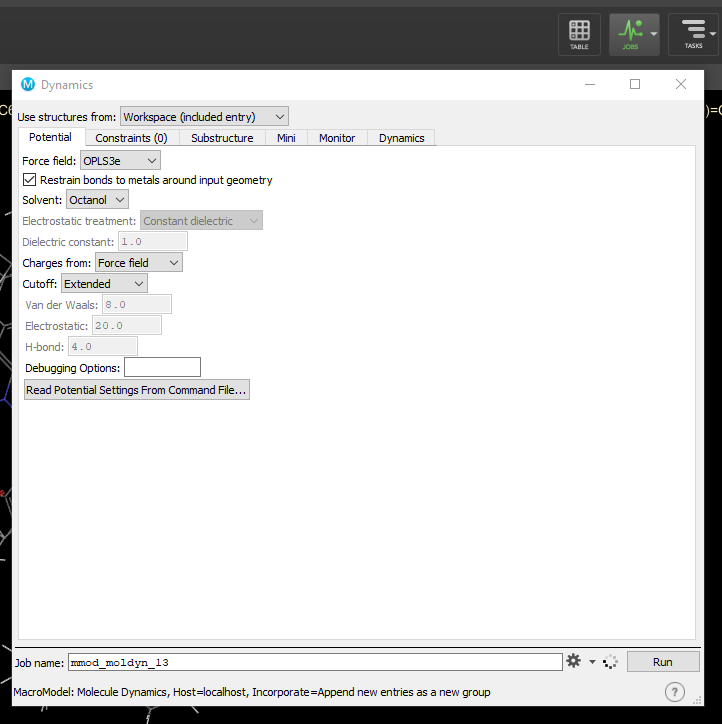
- Go to tasks -> browse -> MacroModel -> Molecular Dynamics to open the dynamics window. Add distance constraints got from NOESY (save them so you can load them later), set number of structures to sample to 10, change simulation time to about 10,000 and click run.
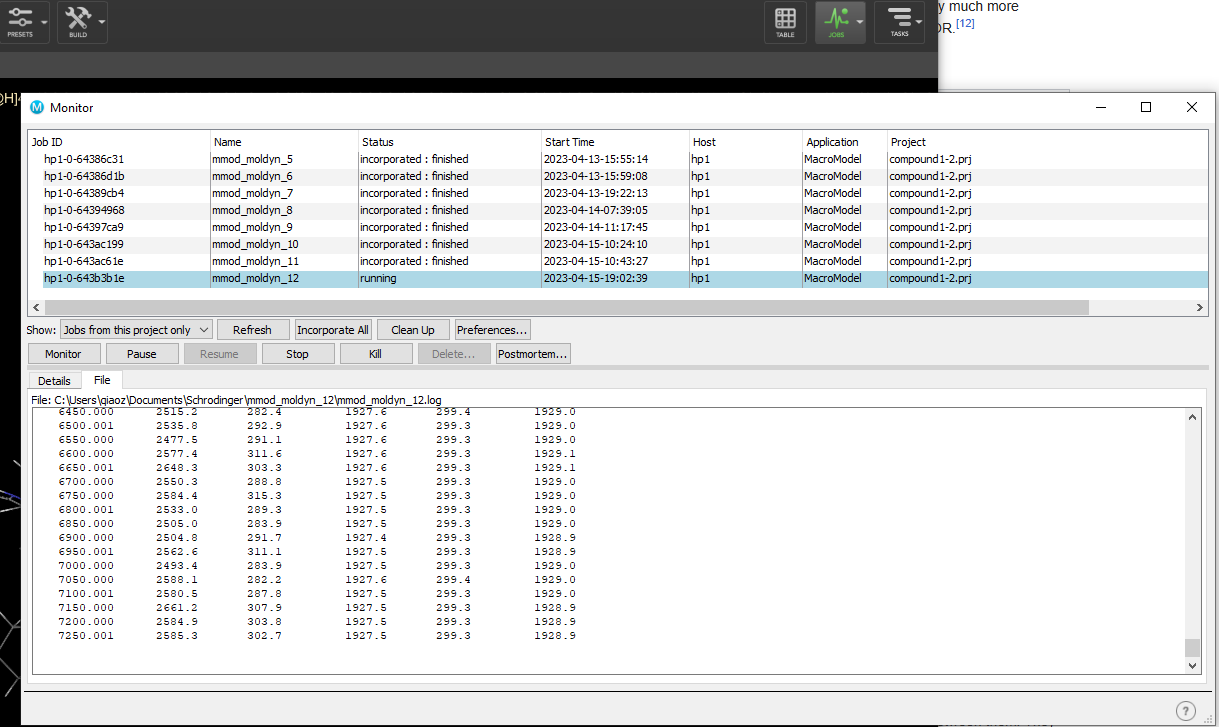
- You can click jobs, monitor to monitor the job status.
- After the job is finished you can load the output file (.maegz) and see the result structure.
Note: Desmond have better molecular dynamics. But it:
- Does not accept any constraints
- The current version (since version 2018) needs GPU and Linux OS.